
La metilación del ADN como ventana al cáncer
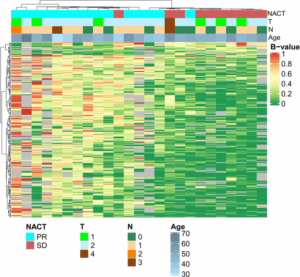 Introducción
Introducción
La oncología molecular, en estas dos décadas, se transformó por completo. Entre los biomarcadores que se han investigado, buscando mejorar la detección, el estudio, y el seguimiento del cáncer, la metilación del ADN surgió como una promesa.
El patrón de metilación del ADN, ósea la adición de grupos metilo a las citosinas en contextos CpG, es un mecanismo epigenético crucial para regular la expresión génica, si. En condiciones normales, esto regula procesos como la diferenciación celular, la inactivación del cromosoma X, y también, el silenciamiento de elementos transponibles.
En el cáncer, con todo, estos patrones se alteran profundamente. Tumores de orígenes muy variados exhiben hipermetilación en promotores de genes supresores de tumores, y, al mismo tiempo, hipometilación global en regiones intergénicas que promueven la inestabilidad genómica.
Es más, esto hace que el perfil de metilación actúe como una firma molecular única, especial para cada tipo de cáncer, e incluso, a veces, para cada paciente.
De lo que era una mera curiosidad epigenética hace dos décadas, hoy se ha convertido en un vibrante campo de diagnóstico no invasivo la biopsia líquida, que además es de medicina personalizada.
Fundamentos biológicos de la metilación del ADN
¿Pero, qué es la metilación exactamente?
Es, en esencia, la agregación de un grupo metilo –CH₃ al carbono 5 de la citosina, dando lugar a la 5-metilcitosina.
Esta reacción ocurre predominantemente en dinucleótidos CpG.
Su función fisiológica es la siguiente:
Silenciar genes cuando la metilación aparece en promotores.
Asegurar la estabilidad genómica.
Influir en la plasticidad celular.
Alteraciones en el cáncer:
Hipermetilación de genes supresores tumorales, por ejemplo, MLH1 en el cáncer colorrectal, y BRCA1 en el cáncer de mama.
Hipometilación global que activan protooncogenes y retrotransposones.
También, hay patrones muy específicos que funcionan como huellas digitales para clasificar los tumores.
Técnicas de análisis de metilación usadas:
En la clínica y en la investigación, se usan diversas metodologías:
La bisulfito-secuenciación, un estándar de oro. Este método transforma las citosinas no metiladas en uracilo, mientras que las metiladas siguen intactas.
Por otro lado, la PCR específica de metilación MSP, resulta ser más accesible, siendo así muy usada en el diagnóstico de rutina en algunos laboratorios.
Arreglos de metilación (Illumina EPIC 850K): Permiten evaluar cientos mil de CpG al mismo tiempo.
Secuenciación de tercera generación (Nanopore, PacBio): Detectan metilación directamente, sin tratamiento químico.
La metilación como biomarcador en oncología…
Aquí es donde esta el gran salto a la práctica clínica:
Detección temprana!
Pruebas como Galleri (GRAIL) emplean perfiles de metilación en ADN libre circulante (cfDNA) para identificar mas de 50 tipos de cáncer, con una muestra de sangre.
Permite detectar tumores, incluso, antes que salgan en pruebas de imagen.
Clasificación del origen tumoral.
Tumores de origen desconocido (TOD) se pueden clasificar de acuerdo a sus huellas epigenéticas.
Esto tiene implicaciones directas, en el tratamiento dirigido.
Pronóstico y predicción de respuesta.
Ciertos patrones predicen resistencia a quimioterapia o respuesta a inmunoterapia.
Ejemplo: Metilación de MGMT en glioblastoma predice respuesta a temozolomida.
Monitoreo de recurrencia.
Un simple análisis de sangre, puede detectar recurrencia, meses antes que las pruebas de imagen tradicionales.
Implicaciones clínicas prácticas
Entrar la metilación del ADN a la clínica, eso sí que cambia el juego:
Verdaderamente medicina personalizada: va más allá del tejido dañado, se mete en la firma molecular de cada persona, ¿sabes?.
Menos procedimientos invasivos: una biopsia líquida basada en cfDNA puede reemplazar muchas biopsias de tejido.
Eficiencia económica: si bien los tests actuales son costosos, a medio plazo bajan los costos asociados a diagnósticos tardíos y tratamientos fallidos, eh.
Ética y psicología médica: detectar el cáncer subclínico temprano trae dudas sobre el sobretratamiento y la ansiedad en los pacientes.
Casos clínicos importantes
Glioblastoma y MGMT
Analizar la metilación de MGMT nos dice si un paciente reaccionará a la temozolomida.
Los que no responden evitan toxicidad innecesaria.
Cáncer colorrectal y MLH1
La metilación de MLH1 señala inestabilidad de microsatélites (MSI), con rol en las terapias.
Biopsia líquida multitumoral (test Galleri)
Pacientes sin síntomas pero con metilación extraña en cfDNA pueden recibir un diagnóstico de cáncer en estadio I.
Retos del presente
Sensibilidad y especificidad: aunque buena, no es perfecta, hay riesgos de falsos positivos y negativos.
Accesibilidad global: Actualmente se limita a centros oncológicos importantes, aquellos ubicados en naciones de altos ingresos.
Estandarización: No existe un acuerdo total sobre qué conjuntos de CpGs deberian emplearse.
Interpretación clínica: Resulta vital diferenciar entre lesiones que son indolentes y las que sí son clínicamente relevantes.
Futuro cercano.
Integración con IA
Algoritmos de aprendizaje profundo; ya clasifican tumores utilizando miles de patrones de metilación.
Pruebas de cribado rutinarias:
En la próxima década, es muy probable que un sencillo análisis sanguíneo, con perfiles de metilación, se integre en los chequeos anuales rutinarios en la población en general.
Medicina preventiva:
La detección temprana de campos precancerosos, antes de que el tumor clínico se manifieste.
📊 Gráficas ilustrativas.
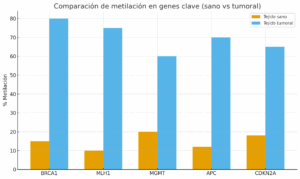
Barras: Los niveles de metilación, en tejido sano vs tejido tumoral.
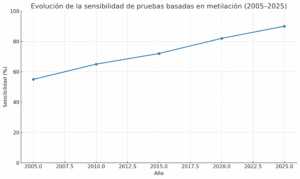
Líneas: la evolución de la sensibilidad de las pruebas de metilación con el transcurso del tiempo (2005–2025).
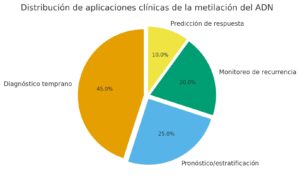
Pastel: La distribución de las aplicaciones clínicas de la metilación diagnósticos pronóstico y seguimiento.
Aquí están los tres gráficos ilustrativos a color:
Barras → comparación de niveles de metilación en genes clave entre tejido sano y tumoral.
Línea → evolución de la sensibilidad, de pruebas basadas en metilación del ADN (2005–2025).
Pastel → distribución de las actuales aplicaciones clínicas de metilación (diagnóstico, pronóstico, monitoreo, y predicción de respuesta).
Interpretar resultados de metilación del ADN no es simple; necesita algoritmos bioinformáticos y estadísticos hechos para reconocer patrones complejos de metilación que separan células sanas y las tumorales, también tipos de cáncer.
📌 Función del algoritmo en la técnica de metilación del ADN
Preprocesamiento de datos, de que.
Los datos crudos de secuenciación o arrays de metilación, tienen millones de señales.
El algoritmo corrige errores técnicos, normaliza intensidades y quita ruido.
Identificación de regiones con metilación diferente (DMRs)
El software busca en genoma, regiones donde la metilación es distinta entre muestra sana y tumoral.
Estas “huellas” son biomarcadores de diagnóstico.
Clasificación con aprendizaje automático
Algoritmos de machine learning, p.ej.
Aprendizaje automático, como los algoritmos de vectores soporte, bosques aleatorios, aprendizaje profundo entrenados sobre conjuntos de datos masivos de pacientes, pueden reconocer firmas de metilación vinculadas a diversos tipos de cáncer.
Un puntaje probabilístico revela si el perfil de metilación sugiere cáncer y, de ser así, su clase específica.
Pronóstico de ubicación tumoral
Mediante la comparación del patrón con atlas como The Cancer Genome Atlas – TCGA, el algoritmo deduce el tejido de origen del cáncer.
Interpretación clínica
El informe final a menudo se expresa clínicamente:
“Probabilidad de detección de cáncer” por ejemplo 92%.
“Posible tejido de origen: colon”.
“Nivel de confianza: alto/medio/bajo”.
📌 Dónde se han originado estos algoritmos
Estados Unidos
La empresa GRAIL, que ahora forma parte de Illumina y que fue posteriormente adquirida por Exact Sciences, creó la prueba Galleri. Este utiliza un algoritmo de aprendizaje automático capacitado con más de 100.000 perfiles de metilación.
El algoritmo se alimenta con datos de cfDNA (ADN circulante libre) para predecir el tipo y el origen del tumor.
Europa
En el laboratorio Europeo de Biología Molecular (EMBL), así como el Wellcome Sanger Institute, del Reino Unido, idearon modelos de IA para la clasificación epigenómica.
El proyecto EPIC array de Illumina y consorcios europeos ayudó a crear algoritmos para los estudios poblacionales, además.
Asia
En China, centros importantes como BGI Genomics y la Universidad de Pekín crearon sistemas propios, utilizando la secuenciación de tercera generación, tipo Nanopore, con algoritmos capaces de detectar la metilación directamente, ¡durante la lectura!
América Latina
Aunque, es menor el alcance, varios grupos en México, Brasil y Argentina se esfuerzan en algoritmos de metilación que se adaptan, con el cáncer cervicouterino y gástrico, pensando en su epidemiología local.
👉 En resumen: Los algoritmos de metilación del ADN son sistemas de bioinformática e inteligencia artificial; ¡transforman patrones epigenéticos en resultados clínicos útiles! Su desarrollo se ha concentrado en Estados Unidos y Europa, con crecimiento en Asia, y poco a poco, comienzan a usarse en Latinoamérica, especialmente para investigaciones concretas.
¿Deseas que te ilustre mediante un diagrama de flujo colorido el funcionamiento, paso a paso, de este algoritmo que va, desde la obtención del cfDNA hasta el informe final clínico?
diagrama de flujo en color, el cual resume el funcionamiento del algoritmo para la interpretación de la metilación del ADN:
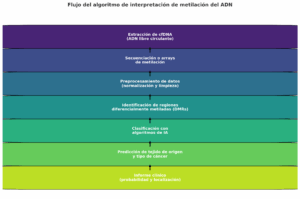
Primero, extraemos el cfDNA de la sangre, cierto.
Luego, viene la secuenciación o los arrays, para conseguir patrones de metilación.
Posteriormente, se lleva a cabo el preprocesamiento de datos, implicando limpieza y normalización.
Después identificamos las regiones metiladas diferencialmente o DMRs, como les llaman.
Clasificamos, valiéndonos de la IA y los algoritmos de machine learning.
Para así predecir el tejido de origen y el tipo de cáncer.
Finalmente, se genera un informe clínico que señala probabilidad y localización.
ACERCA DEL CORRESPONSAL

FRANCISCO JAVIER MARíN MAURI
Me lincencié en psicología por la Universidad de Sevilla. estudios de virología por la Universidad jhons Hopkins y estudios de virus respiratorios emergentes por la O.M.S. Doctorado en neuropsicología por la Universidad de Sevilla. Especialista en Violencia sobre la mujer y en mediación de conflictos sociales.
Llevo desde 1987 ejerciendo la psicología y cada vez pienso más que muchas personas se van de este mundo sin quitarla el sello de fábrica de sus cerebros. Anduve durante casi dos años por varios países africanos para poder realizar mi tesis doctoral sobre el VIH. Ahí aprendes que el poder de la ciencia consiste en tener la suficiente humildad para ejercitar el sentido común que es, por cierto, el menos común de los sentidos.




